My current research in the Specht lab has several facets but center on genome sequencing and assembly, primarily with the Oxford Nanopore. One project looks at convergent evolution of pollinator syndromes in Costus. For this we are assembling genomes for 26 species to investigate genomic changes directly tied to shifts in pollinators. Fine tuning the DNA extraction and cleaning protocols for Nanopore has resulted in getting between 15-20 GB of long-read data on a single flow cell with one library prep.
Myself and many members of the Specht lab are focused on the New World Costus species. Here is an example of the distribution and floral diversity seen in the genus.
Using data from a HybSeq approach that was recently accepted for publication, I was able to look for signatures of selection. Several genes of interest, such as AS2 which is involved in floral fusion, show interesting patterns across the genus.
I am currently working on assembly the genomes of the different species. Using a mostly hybrid approach with MaSuRCA has yielded good but not great results.
After heavy modifications to our DNA preparation steps, I am able to generate 15-20 GB of long-read data per flow cell. This has allowed me to use Flye to assemble with the long-reads only followed by a polishing step. This method has made our assemblies much better and increased our BUSCO genes that we find complete and single copy.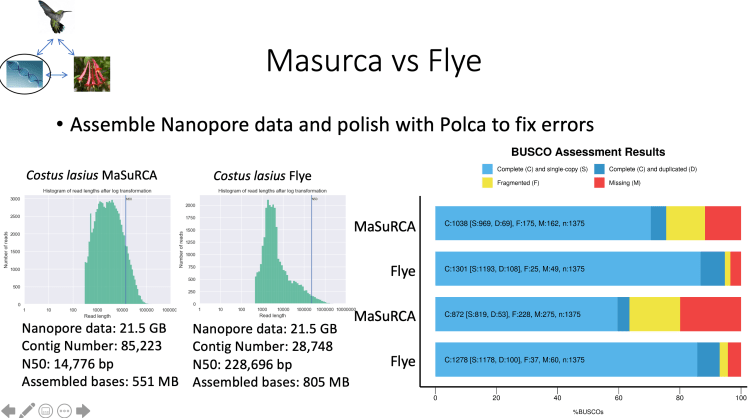
A second project is centered on Calochortus, specifically working on assembling a genome of Calochortus venustus. With a genome of 5.5-6.2 GB, this is no easy feat. The morphological variation in floral forms of Calochortus is fascinating, and we are working on combining genomics, phylogenetics, and cellular morphology to investigate the evolutionary history of this group. Adriana Hernandez, a graduate student in the lab is looking at floral color polymorphisms in C. venustus and I am helping build a draft genome to facilitate SNP calling for population genetic analyses.
Generating a Liliales genome is important based on the evolutionary history of the group. As fo 2019, there are no closely related species that we can leverage a high quality genome from.
A genome almost twice the size of the human genome does present some issues, both in obtaining enough sequencing data for a good assembly to the computational power to carryout the assembly. Here are a few examples of the troubleshooting that I have done to generate a decent draft genome.


Even a fairly poor draft genome can do wonders to help facilitate SNP calling from RAD-Seq data. Here is an example with a subset of data.
Stay tuned for more updates and much better assemblies for both Costus and Calochortus.
